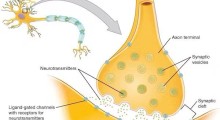
La distruzione di una classe di proteine modificate dallo zucchero migliora la riparazione cellulare, salva la perdita di neuroni e inverte i cambiamenti cellulari associati alle malattie neurodegenerative, secondo dei ricercatori.
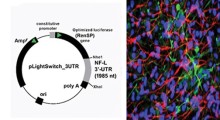
") Due cervelli di moscerino della frutta, marcati per mostrare la morte cellulare, evidenziata dai colori più luminosi. In alto: cervello con deficit nel gene presenilina e molta morte cellulare. In basso: cervello recuperato distruggendo le proteine modificate dal solfato eparan. (Fonte: Alyssa Connell / Selleck Lab / Penn State)
Due cervelli di moscerino della frutta, marcati per mostrare la morte cellulare, evidenziata dai colori più luminosi. In alto: cervello con deficit nel gene presenilina e molta morte cellulare. In basso: cervello recuperato distruggendo le proteine modificate dal solfato eparan. (Fonte: Alyssa Connell / Selleck Lab / Penn State)
Una classe di proteine che regola la riparazione cellulare e migliora i sistemi di segnalazione di crescita cellulare potrebbe essere un nuovo obiettivo promettente per il trattamento del morbo di Alzheimer (MA) e di altre malattie neurodegenerative, secondo un nuovo studio guidato da ricercatori della Penn State.
Essi hanno scoperto che distruggere le modifiche dello zucchero necessarie per queste proteine promuove la riparazione cellulare e inverte le anomalie cellulari che si verificano nelle malattie neurodegenerative. Lo studio è apparso il 2 luglio su iScience e i ricercatori hanno un brevetto relativo a questo lavoro.
"Finora le strategie per trattare il MA si sono concentrate in gran parte su cambiamenti patologici importanti nelle fasi tardive della malattia", ha affermato Scott Selleck, professore di biochimica e biologia molecolare della Penn State e leader del team di ricerca. “Sebbene farmaci approvati di recente dalla FDA abbiano dimostrato la capacità di rallentare modestamente la malattia puntando uno di questi cambiamenti, l'accumulo di amiloide, dei farmaci che puntano i primi deficit cellulari potrebbero fornire strumenti importanti per fermare o invertire la malattia. Siamo interessati a comprendere i primi cambiamenti cellulari che si trovano non solo nel MA, ma che sono condivisi da altre malattie neurodegenerative, come la sclerosi laterale amiotrofica (SLA) e il Parkinson".
Nonostante il suo impatto diffuso, non si conosce la causa o il meccanismo biologico del MA. Le molecole di segnalazione cellulare chiamate 'proteine modificate da eparan solfato' sono state implicate nello sviluppo del MA, ma il loro ruolo specifico è rimasto poco chiaro, ha detto Saleck. In questo studio, il team di ricerca ha prima eseguito una serie di analisi nelle linee cellulari umane e nelle cellule cerebrali di topo che esprimono aspetti del MA, dimostrando che queste proteine regolano i processi cellulari noti per essere colpiti in diverse malattie neurodegenerative.
Le proteine modificate da eparan solfato si trovano sia sulla superficie delle cellule animali che nella matrice tra le cellule. Questa classe di proteine prende il nome da un polimero di zucchero che porta molti gruppi di solfato, chiamato eparan solfato. Le catene di eparan solfato sono attaccate a proteine specifiche e questa modifica consente a queste proteine di assemblare complessi di segnalazione che influenzano la crescita cellulare e influenzano il modo in cui la cellula interagisce con il suo ambiente. Questi percorsi di segnalazione regolano anche l'autofagia, un processo di riparazione cellulare che elimina i componenti danneggiati o disfunzionali nella cellula.
"Nelle prime fasi di diverse malattie neurodegenerative, l'autofagia è compromessa, il che significa che le cellule hanno una capacità ridotta di riparazione", ha affermato Selleck. “In questo studio, abbiamo determinato che le proteine modificate da eparan solfato sopprimono la riparazione cellulare dipendente dall'autofagia. Inoltre, mostriamo che compromettendo la struttura e la funzione delle modificazioni dello zucchero di queste proteine, aumentano i livelli di autofagia, così che le cellule possono occuparsi dei danni".
I ricercatori hanno scoperto che, nelle cellule umane e di topo, ridurre la funzione delle proteine modificate da eparan solfato ha anche recuperato altre patologie che si presentano all'inizio delle malattie neurodegenerative, migliorando la funzione dei mitocondri - che sono responsabili della produzione di energia nella cellula - e riducendo l'accumulo di lipidi o composti grassi, dentro le cellule.
I ricercatori hanno quindi valutato il ruolo delle proteine modificate da eparan solfato in animali modello di MA, moscerini della frutta con deficit in una proteina presenilina. Le mutazioni della presenilina causano una malattia a esordio precoce nell'uomo e anche nei moscerini; la presenilina difettosa provoca morte cellulare e degenerazione cerebrale. Nei moscerini con deficit nella presenilina, ridurre la funzione delle catene di eparan solfato ha soppresso la morte dei neuroni e ha corretto anche altri difetti cellulari. Questi risultati sono direttamente rilevanti per la ricerca sulla genetica umana, hanno affermato i ricercatori.
“Gli individui con mutazioni in un gene presenilina, PSEN1, sviluppano il MA verso i 45 anni, ma se ereditano anche un raro cambiamento genetico in una proteina specifica chiamata ApoE, la malattia è ritardata, a volte di decenni”, ha detto Selleck, spiegando che l'ApoE ha un ruolo importante nel trasporto lipidico e si lega all'eparan solfato. “Questo cambiamento nell'ApoE riduce notevolmente il suo legame all'eparan solfato. Il nostro lavoro parte ed estende questi risultati, implicando direttamente l'eparan solfato nella patologia di MA che coinvolge sia PSEN1 che ApoE. Puntare gli enzimi che producono eparan solfato potrebbe essere un mezzo per bloccare la neurodegenerazione nell'uomo".
Nell'insieme, questi risultati mostrano che interrompere la struttura delle modifiche eparan solfato, blocca o inverte i primi problemi cellulari in questi modelli di MA.
"Salviamo l'animale dalla perdita di neuroni, dai difetti mitocondriali e recuperiamo i deficit del comportamento che fungono da misura della funzione del sistema nervoso", ha affermato Selleck. "Questi risultati suggeriscono un obiettivo promettente per i trattamenti futuri che potrebbero salvare le prime anomalie che si verificano in molte malattie neurodegenerative".
I ricercatori hanno anche esplorato il modo in cui l'espressione genica cambia quando si elimina la capacità delle cellule umane di produrre catene di eparan solfato. Hanno scoperto che si modulano i livelli di espressione di oltre il 50% dei circa 70 geni noti per essere associati al MA ad esordio tardivo, incluso l'ApoE, suggerendo un legame tra proteine modificate da eparan solfato e le forme più comuni e tardive del MA.
"C'è la necessità fondamentale di concentrarsi sui cambiamenti cellulari che si verificano all'inizio della progressione della malattia e di sviluppare trattamenti che li bloccano o li invertono", ha affermato Selleck. “Dimostriamo che autofagia ridotta, difetti mitocondriali e accumulo lipidico - tutti i cambiamenti comuni nelle neurodegenerazioni - possono essere bloccati alterando una classe di proteine, quelle con modifiche da eparan solfato. Pensiamo che queste molecole siano obiettivi promettenti per lo sviluppo di farmaci".
I ricercatori sospettano che interrompere questo percorso per promuovere i sistemi di riparazione cellulare potrebbe essere importante per un'ampia varietà di altre malattie in cui si verificano difetti di autofagia: "Le applicazioni che manipolano questo percorso possono essere ampiamente utili in una serie di condizioni mediche umane", ha affermato Selleck.
Fonte: Gail McCormick in Penn State (> English) - Traduzione di Franco Pellizzari.
Riferimenti: N Schultheis, [+13], SB Selleck. Altering heparan sulfate suppresses cell abnormalities and neuron loss in Drosophila presenilin model of Alzheimer Disease. iScience, 2024, DOI
Copyright: Tutti i diritti di testi o marchi inclusi nell'articolo sono riservati ai rispettivi proprietari.
Liberatoria: Questo articolo non propone terapie o diete; per qualsiasi modifica della propria cura o regime alimentare si consiglia di rivolgersi a un medico o dietologo. Il contenuto non rappresenta necessariamente l'opinione dell'Associazione Alzheimer OdV di Riese Pio X ma solo quella dell'autore citato come "Fonte". I siti terzi raggiungibili da eventuali collegamenti contenuti nell'articolo e/o dagli annunci pubblicitari sono completamente estranei all'Associazione, il loro accesso e uso è a discrezione dell'utente. Liberatoria completa qui.
Nota: L'articolo potrebbe riferire risultati di ricerche mediche, psicologiche, scientifiche o sportive che riflettono lo stato delle conoscenze raggiunte fino alla data della loro pubblicazione.