Un nuovo studio pionieristico guidato da scienziati della University College London e della sanità pubblica GB, ha rivelato, per la prima volta, perché una variante genetica comune peggiora gli esiti di malattia per le persone con SLA e demenza frontotemporale, due malattie neurodegenerative devastanti a insorgenza adulta.
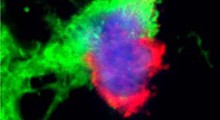
 Aggregazioni anormali della proteina TDP-43, vista nei neuroni colpiti da SLA. Fonte: Alex Bampton / Tammaryn Lashley
Aggregazioni anormali della proteina TDP-43, vista nei neuroni colpiti da SLA. Fonte: Alex Bampton / Tammaryn Lashley
Pubblicato in Nature, lo studio mostra che l'esaurimento della proteina TDP-43, associata a quasi tutti i casi (97%) di sclerosi laterale amiotrofica (SLA) e metà dei casi di demenza frontotemporale (FTD), corrompe le istruzioni genetiche per produrre la proteina neuronale cruciale UNC13A.
Lo studio ha scoperto incredibilmente che una misteriosa variante genetica già associata al rischio di malattia aumenta la possibilità che le istruzioni genetiche dell'UNC13A siano danneggiate nelle persone con le malattie, peggiorando così il rischio e la gravità di SLA e FTD.
L'UNC13A consente ai neuroni (cellule nervose) di comunicare tra loro tramite il rilascio di neurotrasmettitori, e i dati dei modelli animali suggeriscono che la sua perdita dai neuroni può essere fatale. I ricercatori ritengono che la corruzione delle istruzioni genetiche dell'UNC13A nei pazienti possano avere conseguenze altrettanto dannose.
La SLA è la malattia più comune dei neuroni motori e non c'è cura conosciuta; colpisce il cervello e il midollo spinale attaccando i neuroni e i nervi che controllano il movimento, causando la loro morte. Attualmente c'è solo un farmaco approvato per la SLA nel Regno Unito, che estende la durata della vita di pochi mesi ed è efficace solo per una minuscola minoranza di pazienti. Un terzo dei pazienti muore entro un anno dalla diagnosi.
La FTD è una malattia correlata con cause sottostanti simili; i sintomi includono deterioramento del linguaggio, cambiamenti di personalità e difficoltà cognitive.
I ricercatori dicono che la scoperta solleva le speranza di nuovi trattamenti: sviluppando una terapia che blocca la corruzione delle istruzioni genetiche dell'UNC13A, la progressione della malattia potrebbe essere rallentata per la maggior parte delle persone con SLA e circa la metà dei pazienti con FTD.
L'autore senior prof. Pietro Fratta (UCL Queen Square Motor Neuron Disease Centre e Institute of Neurology) ha dichiarato:
"La maggior parte della ricerca sulla terapia genica si è concentrata sui geni implicati nella SLA familiare (pazienti con una storia familiare della malattia), ma la stragrande maggioranza dei casi di SLA è sporadica, senza storia familiare conosciuta".
Il coautore senior dott. Michael Ward (National Institute of Neurological Disorders and Stroke, NIH, Stati Uniti) ha aggiunto:
"Sappiamo da molto tempo che le varianti genetiche nell'UNC13A causano un aumento del rischio di SLA e demenza, ma nessuno aveva capito il perché. Insieme, i nostri team hanno mostrato esattamente il modo in cui questo fattore di rischio genetico della SLA interagisce con il meccanismo di malattia principale, la perdita TDP-43, per peggiorare il corso di malattia".
TDP-43: un giocatore cruciale nella SLA e nella FTD
Probabilmente la proteina più importante nella ricerca sulla SLA è la TDP-43, perché nella maggior parte dei casi (oltre a metà di FTD), la proteina è espulsa erroneamente dal nucleo della cellula. Ciò impedisce alla TDP-43 di eseguire le sue importanti funzioni, come ad esempio garantire che il micro RNA (mRNA, fase intermedia tra DNA e produzione di proteine) sia prodotto correttamente.
Il dott. Ward ha detto:
"Era noto da tempo che la maggior parte dei pazienti con SLA, e circa la metà di quelli con FTD, perdono la funzione di una proteina chiave chiamata TDP-43, provocando il caos nelle cellule nervose che sono interessate. Ma non si sapeva come invertire le conseguenze più problematiche della perdita di TDP-43".
Nell'ambito dello studio sperimentale, i ricercatori hanno usato cellule staminali umane derivate dalla pelle per creare neuroni in un piatto e hanno rimosso la proteina TDP-43 da queste cellule usando una nuova tecnologia basata su CRISPR-CAS9, la tecnologia di modifica dei geni vincitrice del Nobel.
Gli scienziati sono stati quindi in grado di studiare come questi neuroni senza TDP-43 differivano da quelli sani. Hanno scoperto che l'mRNA per la proteina UNC13A era corrotto, il che significa che i ribosomi nei neuroni coltivati in laboratorio non erano in grado di produrre correttamente la proteina UNC13A.
In più, quando la squadra ha esaminato i campioni di cervello di pazienti SLA e FTD, hanno nuovamente trovato che l'mRNA per la UNC13A non era corretto, confermando che i loro esperimenti hanno replicato il processo di malattia del mondo reale.
Dato il ruolo essenziale che la UNC13A ha nella facilitazione della comunicazione dei neuroni, la sua corruzione è quindi suscettibile di compromettere la funzione neuronale e contribuire alla neurodegenerazione in chi ha SLA o FTD.
Le varianti genetiche aumentano il rischio di corruzione dell'mRNA della UNC13A
Il gene dell'UNC13A e le sue proteine corrispondenti sono da tempo sotto la lente dei ricercatori delle malattie dei neuroni motori e FTD, e studi precedenti hanno mostrato che le varianti genetiche comuni aumentano il rischio e la gravità delle malattie, nonostante siano benigne nella maggior parte delle persone non toccate dalle malattie (metà della popolazione porta una di queste varianti, che sono dannose solo nelle persone con SLA o FTD).
Tuttavia, nonostante oltre un decennio di ricerca, l'esatta ragione di questo è rimasta misteriosa, poiché queste varianti non modificano direttamente la sequenza di codifica delle proteine UNC13A, ma si trovano invece in una regione del 'DNA spazzatura'.
I ricercatori ritengono di avere trovato la risposta a questa domanda critica: hanno scoperto che le varianti collegate al rischio aumentano notevolmente le possibilità che l'mRNA delle UNS13A si danneggi quando inizia il corso della SLA e della FTD e la perdita associata della proteina TDP-43. Pertanto, i pazienti con queste varianti genetiche subiscono una maggiore perdita di UNC13A, con una malattia di conseguenza più grave.
Il primo coautore, il dottorando Oscar Wilkins (UCL Queen Square Institute of Neurology e Francis Crick Institute), ha dichiarato:
"Questi risultati rappresentano una svolta significativa per diversi motivi. In primo luogo, spiegano perché le varianti genetiche dell'UNC13A aumentano il rischio di malattie del motoneurone e demenza, una questione che confonde i ricercatori da oltre un decennio. Dimostrano anche per la prima volta un collegamento genetico specifico tra la perdita della funzione della TDP-43 nel nucleo e la SLA, migliorando la comprensione scientifica di questo meccanismo centrale di malattia".
Prossimi passi
Il prof. Fratta ha dichiarato:
"Ci siamo basati su anni di ricerca genetica che ha implicato l'UNC13A nella malattia dei neuroni motori e nella FTD, e l'ha ribadito con una nuova scoperta di biologia molecolare che conferma che il gene è assolutamente fondamentale per il processo di malattia.
"Speriamo nei prossimi anni di portare avanti esperimenti per sviluppare un trattamento che può migliorare, potenzialmente in modo notevole, la vita delle persone con SLA".
I ricercatori sono fiduciosi che, con questa nuova informazione, possono essere create nuove terapie per la malattia dei neuroni motori che impediscano all'mRNA delle UNC13A di essere danneggiato nei pazienti.
Fonte: University College London (> English) - Traduzione di Franco Pellizzari.
Riferimenti: A Brown, ..[+94], Pietro Fratta. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature, 2022, DOI
Copyright: Tutti i diritti di testi o marchi inclusi nell'articolo sono riservati ai rispettivi proprietari.
Liberatoria: Questo articolo non propone terapie o diete; per qualsiasi modifica della propria cura o regime alimentare si consiglia di rivolgersi a un medico o dietologo. Il contenuto non rappresenta necessariamente l'opinione dell'Associazione Alzheimer OdV di Riese Pio X ma solo quella dell'autore citato come "Fonte". I siti terzi raggiungibili da eventuali collegamenti contenuti nell'articolo e/o dagli annunci pubblicitari sono completamente estranei all'Associazione, il loro accesso e uso è a discrezione dell'utente. Liberatoria completa qui.
Nota: L'articolo potrebbe riferire risultati di ricerche mediche, psicologiche, scientifiche o sportive che riflettono lo stato delle conoscenze raggiunte fino alla data della loro pubblicazione.