Un gruppo di ricerca guidato da Susan Ackerman, PhD, professoressa del Jackson Laboratory e ricercatrice «Howard Hughes» ha individuato un meccanismo sorprendente che sta dietro la neurodegenerazione nei topi, che comporta un difetto in un componente chiave del macchinario cellulare che produce le proteine, chiamato «RNA transfer» (tRNA).
I ricercatori riferiscono nella rivista Science che una mutazione di un gene che produce il tRNA, che opera solo nel sistema nervoso centrale, risulta in uno "stallo" o in una pausa del processo di produzione di proteine nei ribosomi neuronali. Quando manca anche un'altra proteina che i ricercatori hanno identificato (GTPBP2), ecco la neurodegenerazione.
"Il nostro studio dimostra che i singoli geni tRNA possono essere espressi specificamente per tessuto nei vertebrati", dice la Ackerman, "e le mutazioni in questi geni possono causare malattie o modificare altri fenotipi. Si tratta di una nuova area per cercare i meccanismi delle malattie".
Sappiamo poco della neurodegenerazione, il processo attraverso il quale i neuroni maturi decadono e infine muoiono, eppure essa sta alla base di gravi malattie umane, come l'Alzheimer, il Parkinson, l'Huntington e la SLA (sclerosi laterale amiotrofica, o malattia di Lou Gehrig). Anche se le cause della neurodegenerazione devono ancora venire alla luce, ci sono sempre più prove che i neuroni siano squisitamente sensibili, molto più di altri tipi di cellule, al deterioramento nella produzione di proteine e del modo in cui si piegano.
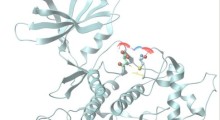
I tRNA sono critici nel tradurre il codice genetico in proteine, i cavalli da soma della cellula. I tRNA possiedono una forma caratteristica a quadrifoglio con due distinti terminali di "lavoro": uno che legge il codice genetico in incrementi di tre lettere (o triplette), e un'altro che trasporta il mattone di costruzione della proteina specificato da ogni tripletta (noto come amminoacido). Negli organismi superiori, i tRNA sono sorprendentemente diversi. Ad esempio, mentre ci sono 61 terzine distinte riconosciute dal tRNA negli esseri umani, il genoma umano contiene circa 500 geni tRNA. Tuttora si sa poco sul motivo per cui sono così numerosi, se hanno funzioni sovrapposte o ridondanti, o se, eventualmente, hanno un ruolo che va oltre la produzione di proteine.
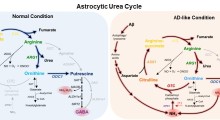
"Diversi geni codificano quasi tutti i tipi di tRNA", dice la Ackerman. "In effetti, i codoni AGA vengono decodificati da cinque tRNA nei topi. Fino ad ora, questa ridondanza apparente ci ha spinto a trascurare completamente il potenziale patogenetico delle mutazioni nel tRNA, così come di altri geni ripetitivi". La Ackerman ed i suoi colleghi del Jackson Laboratory di Bar Harbor nel Maine e di Farmington nel Connecticut, dello Scripps Research Institute di La Jolla in California e della Kumamoto University in Giappone, hanno individuato una mutazione nel gene tRNA n-Tr20 come un colpevole genetico che sta dietro la neurodegenerazione osservata nei topi privi di GTPBP2.
Hanno visto con sorpresa che l'attività del tRNA è confinata nel cervello e in altre parti del sistema nervoso centrale, sia nei topi che nell'uomo. Il tRNA codificato dal n-Tr20 riconosce il codice a triplette AGA (che specifica l'aminoacido arginina). Il difetto n-Tr20 sconvolge il modo di produrre le proteine. In particolare, fa sì che le "fabbriche" responsabili della sintesi delle proteine, chiamate ribosomi, si stallino quando incontrano una tripletta AGA.
Tale stallo può essere in gran parte superato, grazie al lavoro di una proteina associata chiamata GTPBP2. Ma quando questo partner è assente, come succede nel topo mutante che la Ackerman ed i suoi colleghi hanno studiato, lo stallo si intensifica. Questa è ritenuta una forza trainante dietro la neurodegenerazione vista in questi topi.
Fonte: Jackson Laboratory (> English text) - Traduzione di Franco Pellizzari.
Riferimenti: R. Ishimura, G. Nagy, I. Dotu, H. Zhou, X.-L. Yang, P. Schimmel, S. Senju, Y. Nishimura, J. H. Chuang, S. L. Ackerman. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science, 2014; 345 (6195): 455 DOI: 10.1126/science.1249749
Copyright: Tutti i diritti di eventuali testi o marchi citati nell'articolo sono riservati ai rispettivi proprietari.
Liberatoria: Questo articolo non propone terapie o diete; per qualsiasi modifica della propria cura o regime alimentare si consiglia di rivolgersi a un medico o dietologo. Il contenuto non dipende da, nè impegna l'Associazione Alzheimer onlus di Riese Pio X. I siti terzi raggiungibili da eventuali links contenuti nell'articolo e/o dagli annunci pubblicitari sono completamente estranei all'Associazione, il loro accesso e uso è a discrezione dell'utente. Liberatoria completa qui.
Nota: L'articolo potrebbe riferire risultati di ricerche mediche, psicologiche, scientifiche o sportive che riflettono lo stato delle conoscenze raggiunte fino alla data della loro pubblicazione.
| Sostieni l'Associazione; una donazione, anche minima, ci aiuterà ad assistere malati e famiglie e continuare ad informarti. Clicca qui a destra: |