Le malattie che corrono nelle famiglie di solito hanno cause genetiche. Alcune sono mutazioni genetiche che causano direttamente la malattia, se ereditate. Altre dipendono da geni di rischio che influenzano il corpo in modo da aumentare la possibilità che qualcuno svilupperà la malattia.
Nel morbo di Alzheimer (MA), le mutazioni genetiche in uno di 3 geni specifici possono causare la malattia e altri geni di rischio aumentano o diminuiscono il rischio di svilupparla.
Alcune mutazioni o varianti genetiche interagiscono con altre alterazioni genetiche che portano al MA. In alcuni casi, le alterazioni geniche possono interagire con le varianti genetiche che causano il MA in un modo che si rivela benefico; in realtà sopprimono i cambiamenti patologici nel cervello a cui le altre mutazioni porterebbero normalmente.
Queste varianti geniche protettive possono rallentare drasticamente o prevenire il declino cognitivo. In due recenti rapporti di casi di MA familiare, le mutazioni hanno ritardato i sintomi per decenni.
Come neurologo e neuroscienziato, ho trascorso la carriera a studiare il MA e la demenza sia in laboratorio che in clinica. Determinare il modo in cui i geni influenzano la chimica del cervello è vitale per comprendere come progredisce il MA e delineare interventi per prevenire o ritardare il declino cognitivo.
L'ipotesi amiloide
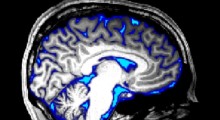
All'inizio degli anni '90, gli scienziati hanno proposto l'ipotesi amiloide per spiegare come si sviluppa il MA. I primi cambiamenti neuropatologici rilevati nel cervello dei pazienti con MA furono la formazione di placche amiloidi, ciuffi di proteine chiamate amiloide-beta (Aβ). Si pensava che altri cambiamenti nel cervello del MA, come l'accumulo di un altro tipo di proteina anormale che formava i grovigli neurofibrillari, si sviluppassero in seguito nel corso della malattia.
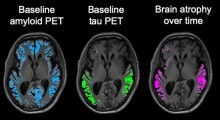
L'Aβ inizia ad accumularsi nel cervello fino a 15 anni prima che emergano i sintomi, che sono correlati al numero di grovigli neurofibrillari nel cervello - più sono i grovigli, peggiore è la cognizione. I ricercatori hanno cercato di determinare se la prevenzione o la rimozione di placche amiloidi dal cervello sarebbe un trattamento efficace.
Immagina l'entusiasmo della comunità scientifica negli anni '90 quando i ricercatori hanno identificato 3 diversi geni che causano il MA familiare, tutti e tre coinvolti con l'Aβ. Il primo era il gene della proteina precursore amiloide. Questo gene induce le cellule a produrre la proteina precursore amiloide, che si rompe in frammenti più piccoli, inclusa l'Aβ che forma placche amiloidi nel cervello.
Il secondo gene è chiamato presenilina 1 (PSEN-1), una proteina necessaria per tagliare la proteina precursore in Aβ. Il terzo gene, la presenilina 2 (PSEN-2), è strettamente correlato al PSEN-1 ma si trova in un numero minore di famiglie con MA familiare.
Queste scoperte hanno aggiunto forza all'ipotesi amiloide, e tuttavia, negli ultimi decenni si sono sviluppate incertezze e opposizioni all'ipotesi amiloide. Ciò è stato in parte legato al riconoscimento che anche molti altri processi - grovigli neurofibrillari, infiammazione e attivazione del sistema immunitario - sono coinvolti nella neurodegenerazione osservata nel MA.
L'ipotesi ha anche avuto un colpo significativo dopo che molti studi clinici, che tentavano di bloccare gli effetti dell'amiloide o di rimuoverla dal cervello, sono falliti. In alcuni casi, i trattamenti avevano effetti collaterali significativi. Alcuni ricercatori hanno escogitato forti difese dell'ipotesi. Ma fino a quando uno studio clinico basato sull'ipotesi amiloide non riesce a dimostrare risultati definitivi, rimane l'incertezza.
Scoperte genetiche con implicazioni terapeutiche
La stragrande maggioranza - oltre il 90% - dei casi di MA si verifica in tarda età, con la prevalenza della malattia che aumenta progressivamente dai 65 anni in su. Tali casi sono per lo più sporadici, senza una chiara storia familiare di MA.
Tuttavia, un numero relativamente piccolo di famiglie ha una delle 3 mutazioni genetiche note per tramandare la malattia. Nel MA familiare, il 50% di ogni generazione erediterà il gene mutato e svilupperà la malattia molto prima, di solito dai 30 a poco dopo i 50 anni.
Nel 2019 e 2023, i ricercatori hanno identificato i cambiamenti in almeno altri due geni che ritardano notevolmente l'insorgenza dei sintomi della malattia nelle persone con mutazioni familiari del MA. Questi geni mutati sono stati trovati in una famiglia molto numerosa in Colombia i cui membri tendevano a sviluppare i sintomi di MA poco dopo i 40 anni.
Una donna della famiglia portatrice di un gene PSEN-1 mutato non ha avuto sintomi cognitivi fino ai 70 anni. Un'analisi genetica ha mostrato che aveva un'ulteriore mutazione in una variante del gene che codifica una proteina chiamata apolipoproteina E (ApoE). I ricercatori ritengono che la mutazione, chiamata variante Christchurch - dal nome dalla città in Nuova Zelanda in cui è stata scoperta la mutazione - è responsabile di interferire e rallentare la malattia.
È importante sottolineare che il suo cervello aveva molte placche amiloidi ma pochissimi grovigli neurofibrillari. Ciò suggerisce che il legame tra i due si era rotto e che il numero soppresso di grovigli neurofibrillari ha anche rallentato la perdita cognitiva.
Nel maggio 2023, i ricercatori hanno riferito che anche due fratelli nella stessa famiglia numerosa non hanno sviluppato problemi di memoria fino ai 60 anni o poco prima degli 80, ed erano portatori di una mutazione in un gene che codifica una proteina chiamata reelina. Gli studi sui topi suggeriscono che la reelina ha effetti protettivi contro la deposizione di placca amiloide nel cervello.
Nel cervello di questi pazienti, come nel paziente che aveva la variante Christchurch, c'erano ampie placche amiloidi ma pochissimi grovigli neurofibrillari. Questa osservazione ha confermato che i grovigli sono responsabili della perdita cognitiva e che ci sono diversi modi per 'disconnettere' l'accumulo di amiloide dai grovigli neurofibrillari.
Dei medicinali capaci di imitare gli effetti protettivi della variante Christchurch o della mutazione reelina potrebbero aiutare a ritardare i sintomi del MA per tutti i pazienti. Poiché la stragrande maggioranza del MA non familiare si manifesta dopo i 70/75 anni, un ritardo di 10 anni nell'emergere dei primi sintomi del MA potrebbe avere un enorme effetto nel ridurre la prevalenza della malattia.
Questi risultati dimostrano che il MA può essere rallentato e si spera che porteranno ad ulteriori nuove terapie che un giorno non solo possono trattare la malattia ma anche prevenirla.
Partenze e fermate
Nonostante oltre 20 anni di dubbi e fallimenti delle terapie, gli ultimi anni hanno visto risultati positivi da tre diversi trattamenti - aducanumab, lecanemab e donanemab - che rimuovono in una certa misura le placche amiloidi e rallentano la perdita della funzione cognitiva. Sebbene vi sia ancora discussione su quanto rallentamento del declino sia clinicamente significativo, questi successi forniscono supporto all'ipotesi amiloide. Suggeriscono anche che saranno necessarie altre strategie per un trattamento ottimale.
L'approvazione del 2021 della FDA del primo trattamento anticorpo per il MA, l'aducanumab, venduto con il marchio Aduhelm, è stata controversa. Solo uno dei due studi clinici che ne testavano sicurezza ed efficacia nelle persone ha prodotto risultati positivi. La FDA ha approvato il farmaco sulla base di quel singolo studio attraverso un processo di approvazione accelerato in cui i trattamenti che soddisfano un'esigenza clinica non soddisfatta possono avere una corsia preferenziale.
Il secondo anticorpo, lecanemab (Leqembi), è stato approvato nel gennaio 2023 attraverso lo stesso percorso di approvazione accelerato e poi approvato completamente a luglio 2023. Il terzo anticorpo, donanemab, ha completato una sperimentazione clinica riuscita di fase 3 e sta aspettando più dati di sicurezza. Quando saranno presentati alla FDA, l'agenzia prenderà in considerazione il farmaco per l'approvazione.
Fonte: Steven Dekosky, professore di neurologia e neuroscienza, Università della Florida
Pubblicato in The Conversation (> English) - Traduzione di Franco Pellizzari.
Copyright: Tutti i diritti di testi o marchi inclusi nell'articolo sono riservati ai rispettivi proprietari.
Liberatoria: Questo articolo non propone terapie o diete; per qualsiasi modifica della propria cura o regime alimentare si consiglia di rivolgersi a un medico o dietologo. Il contenuto non rappresenta necessariamente l'opinione dell'Associazione Alzheimer OdV di Riese Pio X ma solo quella dell'autore citato come "Fonte". I siti terzi raggiungibili da eventuali collegamenti contenuti nell'articolo e/o dagli annunci pubblicitari sono completamente estranei all'Associazione, il loro accesso e uso è a discrezione dell'utente. Liberatoria completa qui.
Nota: L'articolo potrebbe riferire risultati di ricerche mediche, psicologiche, scientifiche o sportive che riflettono lo stato delle conoscenze raggiunte fino alla data della loro pubblicazione.