L'ipotesi 'cascata amiloide' dell'Alzheimer è troppo forte per fallire? (1)
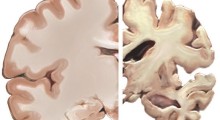
Essa postula che la deposizione nel cervello di proteine amiloide-β è il primo evento cruciale nella patogenesi della malattia, ed è da decenni il fulcro della ricerca sulla demenza (2).
L'ipotesi suggerisce che rimuovendo l'amiloide-β si inverte o si previene l'espressione clinica della demenza.
Tuttavia, in tutti gli studi clinici di fase III eseguiti fino ad oggi, i trattamenti che puntavano l'amiloide-β non sono riusciti a migliorare gli esiti cognitivi, nonostante la riduzione dell'amiloide-β nel cervello (3).
Si sostiene da più parti che questi esperimenti negativi non smentiscono l'ipotesi, ma che i trattamenti devono iniziare decenni prima che amiloide-β 'provochi' la perdita neuronale, o che gli esperimenti debbano includere persone senza depositi di amiloide (2).
I risultati del terzo esperimento sul solanezumab (un anticorpo monoclonale che punta le placche amiloidi) erano quindi molto attesi dopo che i primi due test avevano avuto risultati negativi, ma con indicazioni post-hoc di potenziali effetti sulla "malattia precoce" (4). Lo studio aveva arruolato anziani con una diagnosi clinica di Alzheimer precoce e con depositi di amiloide ed era considerato un 'test importante' dell'ipotesi cascata amiloide (5).
Il risultato negativo dello studio è stato annunciato il 23 novembre 2016 con un tripudio di comunicazione: il solanezumab non ha rallentato il declino cognitivo degli adulti con lieve demenza di Alzheimer. Nonostante questo fallimento di alto profilo, un editoriale su Nature ha sostenuto che il risultato "non uccide la teoria principale della malattia"; al contrario, dobbiamo aspettare gli studi su anticorpi amiloidi più potenti dati prima e l'esito degli altri esperimenti clinici in corso (5).
Questo è vero, naturalmente, ma di solito i ricercatori hanno una profonda comprensione dei meccanismi di base della malattia prima di lanciare studi costosi di fase III con migliaia di partecipanti. Il ruolo esatto dell'amiloide-β nella neurobiologia sottostante la maggior parte delle demenze resta controverso (1, 6, 7).
Nonostante la semplicità dell'ipotesi cascata amiloide la renda seducente, essa sottovaluta gravemente la complessità della scissione della 'proteina precursore dell'amiloide' in amiloide-β e in numerosi altri frammenti proteolitici che potrebbero avere anch'essi un ruolo nella demenza (8, 9).
Nonostante la mancanza di chiarezza sulla specificità di anticorpi cruciali che legano altri peptidi derivati dalla 'proteina precursore dell'amiloide', i risultati della ricerca biomedica sono inevitabilmente interpretati come allineati all'ipotesi prevalente che l'amiloide-β è l'unica proteina neurotossica attiva in questo processo e dovrebbe quindi essere l'obiettivo dell'intervento terapeutico.
Una preoccupazione trascurata, ma importante, è il linguaggio conclusivo usato dai ricercatori di Alzheimer. Ciò deriva dalle risultanze sulle popolazioni più giovani, ma ad alto rischio, con neuropatologie più pure di tipo Alzheimer, piuttosto che sugli anziani (molti ultra novantenni) che rappresentano la maggior parte delle persone con Alzheimer in comunità.
La ricerca nelle popolazioni più giovani, che tende a supportare l'ipotesi amiloide, viene poi traslata in modelli animali e in vitro. Questa a sua volta guida gli sviluppi terapeutici, nonostante il loro uso di progetti [di studio] riduzionisti che controllano strettamente i fattori genetici e ambientali e che tengono conto raramente dell'invecchiamento.
E' stata dimostrata più volte la complessità e la natura mista della demenza negli anziani (10), ma questi risultati rimangono in qualche modo scomodi e scollegati dai grandi investimenti in nuovi interventi.
Gli studi di popolazione dimostrano che le relazioni tra neuropatologia e l'espressione dei sintomi della demenza non sono così deterministiche in età avanzata; la neuropatologia correlata all'amiloide-β può essere grave negli anziani senza demenza e praticamente assente in quelli con demenza (10). Per conciliare queste discordanze, è stato proposto il concetto di 'Alzheimer preclinico'.
Rimane non verificabile l'ipotesi che una persona anziana cognitivamente intatta, con una vasta neuropatologia al momento della morte, avrebbe sviluppato una demenza se fosse vissuta abbastanza a lungo. Sappiamo con certezza, tuttavia, che il legame tra la patologia cerebrale e i sintomi cognitivi è molto variabile nella popolazione umana.
Il fallimento del solanezumab dovrebbe essere un campanello d'allarme per investire in un programma molto più ampio di ricerca sul modo in cui la biologia si collega all'espressione della demenza all'interno di diversi corsi di vita e in età diverse. Si è spesso affermato che la demenza non è una parte del normale invecchiamento, eppure la vecchiaia rimane il fattore di rischio più potente per i sintomi di demenza.
Ci sono analogie tra i cambiamenti biologici osservati nell'Alzheimer e quelli presenti nell'invecchiamento normale (6, 11, 12). Aumentando la nostra comprensione della biologia dell'invecchiamento, e di come manipolarlo, si potrebbero delineare nuovi bersagli terapeutici per l'Alzheimer (13). Il nuovo UK Dementia Research Institute potrebbe dare l'opportunità di concentrarsi su nuovi meccanismi e su persone più anziane (14).
L'approccio 'singola malattia' con interventi altamente specializzati sembra improbabile che sia efficace come può esserlo una serie di altri investimenti che supportano la salute per tutta la vita della popolazione (15), come incoraggiare l'esercizio fisico, una dieta sana, e non fumare e ridurre i farmaci associati ad un aumento del rischio di demenza.
Questo non è un messaggio nuovo, ma non è stato udito abbastanza bene da chi guida lo sforzo globale per prevenire e curare la demenza.
Referenze:
- Castellani RJ, Smith MA. Compounding artefacts with uncertainty, and an amyloid cascade hypothesis that is “too big to fail.” J Pathol 2011;224:147-52. doi:10.1002/path.2885
- Winblad B, Amouyel P, Andrieu S, et al. Defeating Alzheimer’s disease and other dementias: a priority for European science and society. Lancet Neurol 2016;15:455-532. doi:10.1016/S1474-4422(16)00062-4
- Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther 2014;6:37. doi:10.1186/alzrt269
- Siemers ER, Sundell KL, Carlson C, et al. Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement 2016;12:110-20. doi:10.1016/j.jalz.2015.06.1893
- Abbott A, Dolgin E. Failed Alzheimer’s trial does not kill leading theory of disease. Nature 2016;540:15-6. doi:10.1038/nature.2016.21045
- Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 2015;18:794-9. doi:10.1038/nn.4017
- Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun 2014;2:135.pmid:25231068.
- Hunter S, Brayne C. Relationships between the amyloid precursor protein and its various proteolytic fragments and neuronal systems. Alzheimers Res Ther 2012;4:10. doi:10.1186/alzrt108
- Hunter S, Martin S, Brayne C. The APP proteolytic system and its interactions with dynamic networks in Alzheimer’s disease. Methods Mol Biol 2016;1303:71-99. doi:10.1007/978-1-4939-2627-5_3
- Brayne C, Richardson K, Matthews FE, et al. Cambridge City Over-75s Cohort Cc75c Study Neuropathology Collaboration. Neuropathological correlates of dementia in over-80-year-old brain donors from the population-based Cambridge city over-75s cohort (CC75C) study. J Alzheimers Dis 2009;18:645-58.pmid:19661624.
- Hunter S, Arendt T, Brayne C. The senescence hypothesis of disease progression in Alzheimer disease: an integrated matrix of disease pathways for FAD and SAD. Mol Neurobiol 2013;48:556-70. doi:10.1007/s12035-013-8445-3
- Wahl D, Cogger VC, Solon-Biet SM, et al. Nutritional strategies to optimise cognitive function in the aging brain. Ageing Res Rev 2016;31:80-92. doi:10.1016/j.arr.2016.06.006
- Kennedy BK, Berger SL, Brunet A, et al. Geroscience: linking aging to chronic disease. Cell 2014;159:709-13. doi:10.1016/j.cell.2014.10.039
- UCL. New £250m UK Dementia Research Institute to be led from UCL. Press release, 14 Dec 2016. https://www.ucl.ac.uk/news/news-articles/1216/141216-dementia-research-institute
- Brayne C, Davis D. Making Alzheimer’s and dementia research fit for populations. Lancet 2012;380:1441-3. doi:10.1016/S0140-6736(12)61803-0
Fonte: David G Le Couteur, professor of geriatric medicine (Centre for Education and Research on Ageing, Concord Hospital and University of Sydney, Sydney, Australia), Sally Hunter, research associate and Carol Brayne, professor of public health (Cambridge Institute of Public Health, Cambridge, UK)
Pubblicato su: The BMJ (> English text) - Traduzione di Franco Pellizzari.
Copyright: Tutti i diritti di eventuali testi o marchi citati nell'articolo sono riservati ai rispettivi proprietari.
Liberatoria: Questo articolo non propone terapie o diete; per qualsiasi modifica della propria cura o regime alimentare si consiglia di rivolgersi a un medico o dietologo. Il contenuto non dipende da, nè impegna l'Associazione Alzheimer onlus di Riese Pio X. I siti terzi raggiungibili da eventuali links contenuti nell'articolo e/o dagli annunci pubblicitari sono completamente estranei all'Associazione, il loro accesso e uso è a discrezione dell'utente. Liberatoria completa qui.
Nota: L'articolo potrebbe riferire risultati di ricerche mediche, psicologiche, scientifiche o sportive che riflettono lo stato delle conoscenze raggiunte fino alla data della loro pubblicazione.