Scienziati dell'Istituto Scripps Research (TSRI) hanno chiarito uno dei principali meccanismi tossici dell'Alzheimer. Le scoperte potrebbero portare a capire meglio il processo della malattia e a prevenirla.
I risultati, riportati nel numero del 10 Aprile 2013 della rivista Neuron, mostrano che il danno cerebrale nell'Alzheimer è legato all'iperattivazione di un enzima chiamato AMPK (vedi immagine Wikipedia). Quando gli scienziati hanno bloccato questo enzima nei modelli di topo della malattia, i neuroni si sono protetti dalla perdita di sinapsi (punti di connessione da un neurone all'altro) tipica della fase precoce dell'Alzheimer.
"Questi risultati aprono molti nuovi percorsi di studio, compresa la possibilità di sviluppare terapie che colpiscono i meccanismi a monte che portano all'iperattivazione dell'AMPK nel cervello", scrive il professor Franck Polleux del TSRI, che ha guidato il nuovo studio. L'Alzheimer, una malattia neurodegenerativa letale che affligge più di 25 milioni di persone in tutto il mondo, non ha attualmente alcuna cura e nemmeno una terapia per ritardarla.
Oltre alle implicazioni per la scoperta di farmaci per l'Alzheimer, Polleux nota che i risultati suggeriscono la necessità di ulteriori studi per la sicurezza di un farmaco già esistente, la metformina, un trattamento popolare per il diabete di tipo 2, che provoca l'attivazione dell'AMPK.
Indizi allettanti per l'Alzheimer
I ricercatori sanno da anni che le persone nelle prime fasi dell'Alzheimer cominciano a perdere le sinapsi in certe aree del cervello relative alla memoria. Piccoli aggregati di proteina amiloide-beta possono causare la perdita di sinapsi, ma come lo fanno è rimasto finora un mistero. Fino a poco tempo fa, il laboratorio di Polleux era rimasto focalizzato non sulla ricerca di Alzheimer, ma sul normale sviluppo e crescita dei neuroni. Nel 2011, lui ed i suoi colleghi hanno riferito che l'iperattivazione dell'AMPK da parte della metformina, tra gli altri composti, compromette nei modelli animali la capacità dei neuroni di far crescere gli assoni, protuberanze verso l'esterno.
Contemporaneamente, gruppi di ricerca separati hanno trovato indizi che l'AMPK potrebbe avere un ruolo anche nell'Alzheimer. Un gruppo ha riferito che l'AMPK può essere attivato nei neuroni dall'amiloide-beta, fatto che a sua volta può causare una modifica della proteina tau in un processo noto come fosforilazione. Grovigli di tau con diverse fosforilazioni (tau "iperfosforilata") si accumulano nei neuroni nelle aree cerebrali colpite dall'Alzheimer. Questi risultati, pubblicati due anni fa, parlavano di livelli anormalmente elevati di AMPK attivata in questi neuroni infestati da grovigli.
Polleux ha quindi deciso di indagare ulteriormente, per stabilire se le interazioni segnalate, dell'AMPK con l'amiloide-beta e la tau, potessero in effetti causare il danno presente nel cervello dei malati di Alzheimer. "Si sapeva molto poco sulla funzione di questo percorso dell'AMPK nei neuroni, e ci è capitato di avere tutti gli strumenti necessari per lo studio", ha detto.
In cerca di risposte
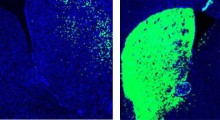
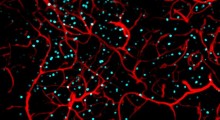
La maggior parte degli esperimenti del nuovo studio sono stati eseguiti da Georges Mairet-Coello, un socio di ricerca post-dottorato nel laboratorio di Polleux. Ha iniziato dalla conferma che l'amiloide-beta, sotto forma di piccoli aggregati ("oligomeri") tossici per le sinapsi, fa effettivamente attivare fortemente l'AMPK; gli oligomeri di amiloide-beta stimolano alcuni recettori neuronali, che a loro volta provocano un afflusso di ioni di calcio nei neuroni. Ha scoperto che questo afflusso del calcio avvia l'attivazione di un enzima chiamato CAMKK2, che sembra essere il principale attivatore dell'AMPK nei neuroni.
Il team ha poi dimostrato che questa sovrattivazione dell'AMPK nei neuroni è la ragione fondamentale dell'effetto dannoso dell'amiloide-beta per le sinapsi. Di solito l'aggiunta di oligomeri di amiloide-beta ad una cultura di neuroni provoca la rapida scomparsa di molte delle spine dendritiche dei neuroni (gambi, tipo radici, attaccati alle sinapsi) che ricevono segnali da altri neuroni. Con una serie di test, gli scienziati hanno dimostrato che gli oligomeri di amiloide-beta non possono causare questa perdita delle spine dendritiche a meno che non si verifichi l'iperattivazione dell'AMPK, e che addirittura l'iperattivazione dell'AMPK, da sola, può causare la perdita delle spine.
Per un esperimento cruciale il team ha utilizzato topi J20, che sono geneticamente modificati per produrre un eccesso di amiloide-beta mutante, e, infine, sviluppare una condizione di tipo Alzheimer. "Quando i topi J20 hanno solo tre mesi di vita, mostrano già una forte diminuzione della densità delle spine dendritiche, in un insieme di neuroni relativi alla memoria che sono anche colpiti presto nell'Alzheimer umano", scrive Mairet-Coello. "Ma quando abbiamo bloccato l'attività del CAMKK2 o dell'AMPK in questi neuroni, abbiamo completamente impedito la perdita di spine".
Successivamente Mairet-Coello ha studiato il ruolo della proteina tau. Di solito serve da elemento strutturale negli assoni neuronali, ma nell'Alzheimer diventa in qualche modo iperfosforilata e si accumula in altre aree neuronali, tra le quali i dendriti, dove la sua presenza è associata alla perdita delle spine. Recenti studi hanno dimostrato che la tossicità dell'amiloide-beta per le spine dendritiche dipende in gran parte dalla presenza della tau, ma non era chiaro come interagiscono le due proteine di Alzheimer.
Il team ha preso spunto da uno studio sui moscerini della frutta (Drosophila) del 2004, in cui una fosforilazione dell'enzima di tipo AMPK in siti specifici sulla proteina tau portava ad una cascata di ulteriori fosforilazioni e alla degenerazione delle cellule nervose. Gli scienziati hanno confermato che uno di questi siti (S262) è in effetti fosforilato dall'AMPK. Essi hanno poi dimostrato che questa fosforilazione specifica della tau costituisce una parte significativa della tossicità sulle sinapsi da parte dell'amiloide-beta. "Il blocco della fosforilazione nel S262, con una forma mutante di tau che non può essere fosforilata in quel sito, ha impedito l'effetto tossico dell'amiloide-beta sulla densità di spine", riferisce Mairet-Coello.
Il risultato suggerisce che l'amiloide-beta contribuisce all'Alzheimer attraverso l'AMPK, per lo più come facilitatore di tossicità della tau.
Altri studi davanti
Mairet-Coello, Polleux ed i loro colleghi stanno andando avanti con ulteriori esperimenti per determinare quali altri processi tossici, come l'eccessiva autofagia, siano promosse dall'iperattivazione dell'AMPK e che potrebbero anche contribuire agli aspetti a lungo termine della progressione dell'Alzheimer. Sono anche interessati agli effetti a lungo termine del blocco dell'iperattivazione dell'AMPK del modello J20 di topo, così come di altri modelli di topo di Alzheimer, che normalmente sviluppano deficit cognitivi in fasi successive. "Abbiamo già contatti nel settore farmaceutico, potenzialmente interessati a colpire il CAMKK2 o l'AMPK", dice Polleux.
Gli altri partecipanti allo studio, finanziato in parte dal National Institutes of Health e dalla Novartis, sono Julien Courchet, Simon Pieraut, Virginie Courchet e Anton Maximov, tutti del TSRI.
Fonte: Scripps Research Institute.
Riferimento: Georges Mairet-Coello, Julien Courchet, Simon Pieraut, Virginie Courchet, Anton Maximov, Franck Polleux. The CAMKK2-AMPK Kinase Pathway Mediates the Synaptotoxic Effects of Aβ Oligomers through Tau Phosphorylation. Neuron, 2013 DOI: 10.1016/j.neuron.2013.02.003.
Pubblicato in Science Daily il 10 Aprile 2013 - Traduzione di Franco Pellizzari.
Copyright: Tutti i diritti di eventuali testi o marchi citati nell'articolo sono riservati ai rispettivi proprietari.
Liberatoria: Questo articolo non propone terapie o diete; per qualsiasi modifica della propria cura o regime alimentare si consiglia di rivolgersi a un medico o dietologo. Il contenuto non dipende da, nè impegna l'Associazione Alzheimer onlus di Riese Pio X. I siti terzi raggiungibili da eventuali links contenuti nell'articolo e/o dagli annunci pubblicitari proposti da Google sono completamente estranei all'Associazione, il loro accesso e uso è a discrezione dell'utente. Liberatoria completa qui.
Nota: L'articolo potrebbe riferire risultati di ricerche mediche, psicologiche, scientifiche o sportive che riflettono lo stato delle conoscenze raggiunte fino alla data della loro pubblicazione.
| Sostieni l'Associazione; una donazione, anche minima, ci aiuterà ad assistere malati e famiglie e continuare ad informarti. Clicca qui a destra: |