Grafica: Galen Dara / American Scientific
Grafica: Galen Dara / American Scientific
Nessun ostacolo fondamentale ci impedisce di sviluppare un trattamento efficace per il morbo di Alzheimer (MA). Altri problemi della natura umana, come la violenza, l'avidità e l'intolleranza, hanno una sconcertante varietà di cause scoraggianti e di incertezze. Ma il MA, al suo interno, è un problema di biologia cellulare la cui soluzione dovrebbe essere alla nostra portata.
C'è una discreta possibilità che la comunità scientifica possa già avere un trattamento riconosciuto, nascosto in un congelatore di laboratorio tra numerose fiale di sostanze chimiche. E in questo momento possono esserci grandi informazioni in attesa di essere notate, nelle grandi banche dati o nei registri delle cartelle cliniche, dei profili neuropsicologici, negli studi di scansione cerebrale, nei marcatori biologici nel sangue e nel liquido spinale, nei genomi, nelle analisi di proteine, nelle registrazioni dei neuroni o nei modelli di colture cellulari e animali.
Ma finora abbiamo perso quegli indizi perché per decenni abbiamo passato troppo tempo alla caccia di ogni nuova scoperta vistosa nella ricerca del MA e troppo poco tempo a pensare profondamente alla biologia alla base di questo disturbo. Invece il nostro lavoro è stato guidato da una serie di ipotesi. Tra questi assunti c'era il ruolo centrale e dominante del frammento della proteina chiamata amiloide-beta (Aβ). Una grande quantità di dati sostiene l'idea che l'Aβ ha un ruolo importante nella malattia. Abbiamo farmaci già sviluppati che possono ridurre la concentrazione dei frammenti di proteine nelle persone affette da MA, ma nel complesso non hanno evitato in modo significativo il declino cognitivo dei pazienti.
Ora sembra semplicistico concludere che l'eliminazione o l'inibizione dell'Aβ può curare o trattare coloro che soffrono di questa malattia, soprattutto senza conoscere molto più profondamente e completamente come si sviluppa e progredisce. Non abbiamo solo abbaiato contro un albero di ricerca completamente sbagliato, ma il nostro zelo ci ha portato ad ignorare altri alberi e persino le radici di questa pianta particolare.
È tempo di tornare alle origini. Sono uno scienziato coinvolto da tre decenni nella ricerca di MA, ho fatto parte di grandi progetti che indagano le famiglie con un elevato rischio di MA, le strategie di prevenzione e la fisiologia dei danni alle cellule cerebrali che fanno parte della malattia. Io e i miei colleghi, che lavorano in molte discipline scientifiche e mediche, crediamo di aver bisogno di riesaminare la fisiologia e la biologia fondamentale del MA, così come rivalutare il contenuto dei database e dei nostri frigoriferi di laboratorio per gli indizi che forse abbiamo trascurato. Questo approccio ci permetterà di sviluppare teorie e modelli del modo in cui progredisce questa malattia, e possiamo usare quelle idee per creare nuove strategie per combattere la malattia.
Ci sono almeno cinque direzioni, aree di ricerca potenzialmente fruttuose e tempestive basate su scoperte importanti fatte negli ultimi anni, che possono estendere la nostra conoscenza, e credo che abbiano una buona probabilità di produrre le intuizioni necessarie per trovare trattamenti efficaci. Queste aree vanno dal malfunzionamento nel modo in cui le cellule del cervello si sbarazzano delle proteine-problema, ai danni causati dall'infiammazione, ai problemi nei modi in cui le cellule inviano segnali elettrici l'una all'altra. Si tratta di domini diversi, ma in una persona si sovrappongono per creare la malattia nel cervello, e singolarmente o in tandem possono trovarsi dietro il danno terribile fatto dal MA.
1. Problemi con lo smaltimento delle proteine
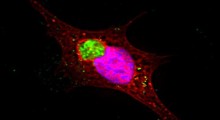
A partire dagli inizi del 1900, diversi neuropatologi, tra cui Alois Alzheimer, lo scienziato dal quale la malattia prende il nome, hanno descritto le lesioni microscopiche nel cervello dei pazienti che erano morti con varie forme di demenza. Oggi sappiamo che questi sono grumi di proteine dalla forma alterata. Nel caso del MA, alcuni dei grumi sono costituiti da pezzi di proteina Aβ. Si posizionano tra i neuroni e sono chiamati placche senili. Altri grumi risiedono all'interno dei neuroni, composti dalla proteina tau, e sono chiamati grovigli neurofibrillari.
Quello che ancora non sappiamo, più di un secolo dopo, è il motivo per cui le cellule non riescono a rimuovere questi grumi anormali. I meccanismi cellulari della rimozione delle proteine danneggiate sono antichi come la vita stessa. Cosa va male nel caso del MA? Questa domanda è centrale per il processo della malattia proprio come la perdita di controllo sulla proliferazione cellulare lo è per la progressione del cancro.
Alcune osservazioni recenti di ricercatori dell'Università di Washington di St. Louis, tra le altre istituzioni, indicano che le proteine anomali possono trovare una loro via d'uscita dalle cellule, eludendo forse i loro sistemi di rilevamento naturale delle molecole cattive. Noi non sappiamo come lo fanno, ma capirlo potrebbe essere un modo molto utile per avviare una nuova ricerca di come e perché il MA progredisce.
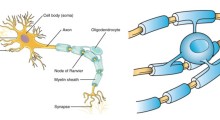
Le cellule hanno due sistemi principali per la rimozione delle proteine anomali: il sistema ubiquitina-proteasoma (UPS) e l'autofagia. Nel primo caso, le proteine sono inserite in una struttura cellulare a forma di botte, chiamata proteasoma, dove vengono masticate in parti riutilizzabili; nella seconda, la cella avvolge le proteine aberranti e le distrugge totalmente. Nei neuroni, questi sistemi sono co-optati per controllare la composizione delle connessioni della segnalazione cellulare, (formate da strutture anatomiche chiamate assoni, dendriti e sinapsi), rafforzandole o indebolendole durante l'apprendimento. (A volte i neuroni danneggiati espellono le proteine e per la loro distruzione si affidano alle microglia, cellule cerebrali che fanno parte del sistema immunitario).
La decisione sull'opportunità di portare una proteina anomala verso l'UPS o autofagia si basa principalmente sulla dimensione della proteina. Il proteasoma ha un'apertura stretta, tipo pori, ad ogni estremità, in grado di accettare un filamento proteico piccolo, raffinato, filiforme. All'interno ci sono enzimi che degradano le proteine nei suoi amminoacidi costituenti, che vengono riciclati per sintetizzare nuove proteine. Le molecole più grandi che non entrano nel proteasoma, come i grumi di proteine e quelle vecchie edeformi con danni legati all'età, vengono traghettate verso il sistema dell'autofagia e il suo motore più potente di distruzione, il lisosoma.
Nel nel MA qualcosa va male, e lascia le cellule cerebrali con questi pezzi di tau e Aβ che le danneggiano ulteriormente e le soffocano. Così potremmo capire molto della patologia del MA, se capissimo i dettagli di questi sistemi. Dobbiamo esaminare differenze specifiche nelle vie di degradazione in diversi sottotipi di neuroni, nonché il preciso meccanismo con cui questi sistemi di smaltimento riconoscono le proteine anomale. Le malformazioni nelle proteine come la tau non avvengono in un unico passaggio.
Le proteine possono ospitare mutazioni e modifiche accumulate che le predispongono al misfolding (errata piegatura), che può essere seguita da aggregazione in strutture più grandi in un processo multistadio. Mentre le proteine progrediscono lungo questo percorso, a che punto entrano in ballo i sistemi di sorveglianza e le riconoscono come anomale? Una conoscenza approfondita di questi tipi di processi ci potrebbe portare ad un approccio più strategico per il trattamento e l'intervento con farmaci.
Una scoperta intrigante che influenza la nostra comprensione di questa evasione è che la tau può viaggiare fuori delle cellule e negli spazi tra di loro, e da lì viene assorbita dalle cellule vicine. Lo scopo di questo sistema di transito è sconosciuto. Lo scambio della proteina tra le cellule è normale, o le cellule scaricano la tau anomala per liberarsi di una sostanza tossica? Pensiamo che nel MA, almeno alcune delle proteine tau fuori delle cellule siano già mal ripiegate. Lo crediamo perché quando tale tau entra in una cellula vicina, forma un modello, un modello anomalo, che altre proteine tau in quella cellula usano per conformare se stesse in modo altrettanto strano. Quando si diffonde, la tau nelle cellule vicine copia la forma specifica della proteina tau in entrata.
Le osservazioni della tau al di fuori delle cellule hanno indotto alcuni a ipotizzare che la proteina potrebbe essere intercettata ed eliminata in quel punto da un anticorpo erogato al paziente. Ma questo approccio è improbabile che funzioni, a meno che non sappiamo esattamente come è deformata la tau quando fa il suo danno. Questa struttura precisa è l'informazione necessaria per progettare un anticorpo altamente specifico. Un'altra questione aperta è dove risiede la tau nello spazio complesso tra le cellule.
Più in particolare, si muove tra le sinapsi, dove due neuroni trasmettono i loro segnali? Questo spazio sinaptico è una stretta fessura che non è facilmente accessibile per un anticorpo. Approcci potenzialmente più promettenti sono capire esattamente come la tau viene estrusa dalle cellule e i recettori che le cellule vicine usano per portare dentro la proteina; recenti esperimenti nel mio laboratorio possono indicare l'identità di uno di questi recettori.
2. Identificare i cambiamenti alle proteine
Un progresso recente importante nella ricerca di MA è la scansione della tau anomala all'interno di una cellula, aggrovigliata in ammassi neurofibrillari, ad un livello di dettaglio mai visto prima. Questa immagine notevole, pubblicata nel 2017 su Nature, ha mostrato migliaia di proteine tau allineate come coppie ermeticamente chiuse in una configurazione a C. È possibile che le caratteristiche viste in questa inclusione solida possano fornire le informazioni necessarie per progettare piccole molecole che si inseriscono nelle fessure della proteina anormale e la separino interrompendo il processo della malattia.
Ma rompere queste strutture è un obiettivo impegnativo per molti motivi, non ultimo la forza con cui l'intero groviglio è tenuto insieme. Una direzione più efficace potrebbe essere quella di determinare la sequenza di eventi microscopici che portano queste proteine tau dal loro stato tipico di tipo liquido a quello più rigido e solido visto in quell'immagine, e scoprire le modificazioni della proteina tau che predispongono a questo cambiamento.
Il passaggio da liquido a solido è chiamato 'transizione di fase'. L'interesse dei biologi in tali transizioni nelle cellule viventi è ora in aumento a causa del loro possibile ruolo nella malattia. I fisico-chimici studiano da molti anni la separazione di fase, come la condensazione di gocce di olio nell'acqua. Olio e acqua sono entrambi liquidi, tuttavia rimangono separati a causa di un equilibrio di forze attraenti e repellenti. Il vantaggio della separazione di fase per le cellule viventi è che concentra un insieme specifico di molecole in un unico luogo, aiutando alcune attività cellulari.
Proteine multiple vicine a un gene, per esempio, possono condensarsi per controllare l'espressione di quel gene, come mostrato da uno studio del 2018 su Science. Un tale set abbreviato di proteine, anche se ancora allo stato liquido, non si diffonde; sono tenute insieme come una gocciolina da forze fisiche deboli. Questa configurazione permette a insiemi di proteine di muoversi e lavorare insieme senza essere piegate in una membrana, che richiederebbe manutenzione con risorse costose dalla cellula.
Alcune proteine, come la tau, quando si trovano all'interno di una goccia sono ammassate, e le concentrazioni elevate potrebbero renderle inclini ad aggregarsi in un groviglio. Le proteine che formano goccioline in questo modo condividono una proprietà nota come 'disordine intrinseco'. Come il dio greco Proteo, possono assumere numerose forme, in contrasto con le proteine più ordinate che sono limitate ad alcune forme specifiche. Diverse forme richiedono diversi livelli di energia. A volte, alcune proteine intrinsecamente disordinate si piegano in un tale stato di bassa energia che non possono spostarsi fuori di esso, aumentando sostanzialmente la loro rigidità. E ciò può aggravare la loro tendenza a aggrovigliarsi insieme.
Le cellule impacchettano anche proteine e altre molecole soggette a transizioni di fase in organelli senza membrana detti 'granuli di stress' e 'granuli di RNA'. Quando certe proteine e RNA si fondono in tali granuli, si impacchettano strettamente insieme ma di solito rimangono in uno stato liquido. Ad una certa densità, tuttavia, possono diventare predisposte a maggiore aggregazione e ad un cambiamento di fase verso quella solida, un cambiamento che farebbe aumentare la loro capacità di causare danni al cervello e le renderebbe più difficili da rimuovere per i sistemi cellulari di smaltimento. Ecco perché dobbiamo capire meglio le condizioni che attivano questo processo.
3. L'influenza dei geni
Nelle persone di mezza età, il MA può derivare da mutazioni genetiche in tre geni (APP, PSEN1 e PSEN2) che causano una forma familiare rara della malattia, un'eredità spaventosa passata da una generazione a quella successiva. Ma per la stragrande maggioranza dei casi, il MA si mostra in soggetti di età superiore a 65 ANNI e non coinvolge questi geni. Selezionando tra decine di migliaia di genomi, i genetisti hanno scoperto altri cambiamenti del DNA, circa due dozzine di varianti genetiche, che fanno aumentare di poco il rischio.
La più influente di queste forme alternative è una versione del gene APOE chiamata variante E4. Una combinazione di diverse varianti genetiche di rischio aumenta la propria probabilità di contrarre la malattia. (Poiché le varianti genetiche spesso sono associate con l'etnia, abbiamo bisogno di un insieme di dati molto più inclusivi, rispetto alle analisi genetiche per lo più basate sull'etnia Caucasica, e ai registri attualmente disponibili, per effettuare una valutazione attendibile del rischio genetico in tutte le popolazioni).
Ognuna di queste varianti apre una porta diversa attraverso la quale possiamo esplorare i modi in cui un piccolo cambiamento nel nostro genoma può aumentare la probabilità di contrarre il MA. Alcune delle varianti viste con più frequenza, e quindi le porte più interessanti, sono geni o altri tratti del DNA nelle microglia. In una ricerca pubblicata su Science nel 2019, che ha esaminato queste cellule del sistema immunitario, gli scienziati hanno scoperto una variante associata al rischio di MA nel gene BIN1. Questo gene è di norma coinvolto nel modo in cui le microglia fagocitano le molecole esterne potenzialmente dannose e le spostano dentro la cellula, proteggendo i neuroni vicini. La variante può influire sull'efficacia con cui le microglia ripuliscono le proteine randage.
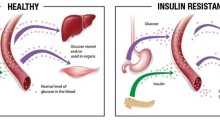
Nelle microglia e in altre cellule, alcune varianti genetiche sono associate anche con l'età e il sesso. Esistono differenze tra uomini e donne, per esempio, per i geni su 22 coppie di cromosomi non sessuali e per i geni espressi sui cromosomi X e Y. Gli effetti di queste varianti possono avere qualcosa a che fare con i tassi più alti di MA nelle donne, che restano anche dopo la correzione per la maggiore longevità delle donne. Nel complesso i piccoli effetti di ogni singola variante genetica associata con il MA, probabilmente contribuiscono, ciascuna con il suo modo limitato, alle differenze individuali nel modo in cui gestiamo l'accumulo di Aβ e tau. Abbiamo bisogno di fissare il come e il perché di questi contributi.
4. Domare l'infiammazione
Quando il cervello rileva una fonte di danno, come le placche amiloidi o i grovigli neurofibrillari tau, suona un allarme e rilascia una raffica di molecole del sistema immunitario chiamate citochine, insieme a varie cellule di attacco. Questa risposta deriva dalle microglia, in gran parte, e provoca una reazione infiammatoria destinata a distruggere qualsiasi tessuto che ospita i punti nei guai. Questo sistema di forza bruta 'innato' opera in modo diverso da quello più raffinato del sistema immunitario 'adattativo', che genera cellule immunitarie e anticorpi che reagiscono solo ad invasori specifici, come batteri o virus, e che montano una risposta di difesa più stretta e precisa.
La risposta innata ampia domina nel MA. Mentre le lesioni proliferano al di là della capacità dei meccanismi interni di un neurone di sbarazzarsi dei detriti, interviene questa risposta generale infiammatoria e, purtroppo, spesso colpisce anche le cellule sane del cervello. Gli scienziati della University of California di Irvine, di recente hanno scoperto che eliminare le microglia invecchiate nei topi anziani ha spinto gli animali a ripopolare il loro cervello con microglia fresche. Questo ringiovanimento ha migliorato la memoria spaziale, invertito i cambiamenti legati all'età nell'espressione genica neuronale, e ha aumentato la nascita di nuovi neuroni, così come la densità dei loro dendriti.
Questa aggressione scatenata da Aβ e tau probabilmente si aggiunge a un livello basso di infiammazione nel cervello che avviene naturalmente con l'invecchiamento. Molti anziani hanno concentrazioni più elevate di citochine proinfiammatorie, come il 'fattore di necrosi tumorale', suggerendo che un lieve stato infiammatorio esiste in tutto il corpo a questo punto della vita. L'invecchiamento è molto variabile tra gli esseri umani, e le differenze implicano che pure il progresso e gli effetti del MA sono molto variabili. Alcune di questa diversità sono probabilmente da attribuire alla variazione individuale nel sistema immunitario umano.
Persone diverse ereditano configurazioni diverse dei geni coinvolti nella risposta immunitaria. Inoltre, durante la vita i nostri sistemi sono modellati da influenze non ereditabili. Siamo soggetti a esposizioni diverse ai microbi simbiotici in luoghi come il nostro intestino e ai microbi patogeni dal nostro ambiente. Tutto questo suggerisce che l'esposizione del sistema immunitario a vari agenti patogeni, così come le nostre differenze genetiche, possono contribuire al modo in cui il MA si sviluppa, attraverso la definizione di un profilo immunitario individuale, o “immunotipo”.
La sfida per i ricercatori che vogliono fermare il danno cerebrale causato dall'infiammazione diffusa è distinguere le risposte immunitarie desiderabili che il cervello usa per combattere lo sviluppo di problemi e il degrado ordinario indotto dall'età, dalle altre risposte immunitarie più spericolate alla patologia del MA in avanzamento. La comunità di ricerca vorrebbe domare l'infiammazione del cervello causata dalla malattia, ma non sa ancora come intervenire con precisione.
5. Disconnessioni elettriche
Il cervello è un organo elettrico: la caratteristica più definente è la sua capacità di codificare e trasmettere le informazioni sotto forma di segnali elettrici passati tra i neuroni, di solito da sostanze chimiche chiamate neurotrasmettitori. È stato già sufficientemente studiato come il MA deteriora la segnalazione delle cellule del cervello e distrugge il loro modo di assemblarsi in circuiti di memoria funzionali. Ma ora è fiorente la capacità di rilevare sia i collegamenti strutturali che funzionali grazie ai progressi tecnici che ci permettono di visualizzare tali collegamenti in dettaglio squisito.
Alcuni di questi progressi coinvolgono l'optogenetica, un modo con cui gli scienziati possono stimolare neuroni specifici nel cervello di un animale con la luce. I ricercatori sono in grado di offrire all'animale una ricompensa o un'esperienza spaventosa, quindi individuare quali geni diventano più attivi. Questo approccio, una conquista impressionante, ora consente ai ricercatori di osservare e manipolare neuroni specifici che codificano una memoria specifica nota come 'engramma', come osservato in uno studio su Science del 2020. Quando queste cellule venivano stimolate solo dalla luce dopo l'esperienza iniziale, ne veniva richiamato il ricordo. Se saremo in grado di capire la biologia che guida la formazione di queste connessioni elettriche della memoria, quell'informazione sarà cruciale per aiutarci a capire come il MA interrompe questo circuito neurale.
I neuroscienziati hanno fatto un altro progresso quest'anno, quando hanno scoperto che le microglia sembrano essere coinvolte nell'indurre il cervello a dimenticare questi engrammi eliminando le sinapsi che normalmente connettono i neuroni.
Sappiamo anche che i neurotrasmettitori sono influenzati in modo diverso da alcune delle proteine coinvolte nella patologia del MA. La tau, per esempio, si accumula nei neuroni che usano il neurotrasmettitore glutammato e lavora per eccitare i segnali. Ma altri neuroni che inibiscono i segnali (la segnalazione si basa su buoni meccanismi di inizio-e-arresto) rilasciano un neurotrasmettitore differente, il GABA, e sono meno influenzati dall'accumulo di tau. La base di questa selettività cellulare e le sue conseguenze è sconosciuta, e dobbiamo capire molto meglio. Gli scienziati hanno anche visto che l'attività neuronale aumenta la diffusione di tau, che potrebbe essere un'altra parte importante del puzzle del MA.
Non solo la segnalazione di vari tipi di cellule è influenzata diversamente dal processo della malattia, ma gli effetti variano anche in aree diverse del cervello. Ad esempio, le aree del cervello legate alla memoria, alle emozioni e al sonno sono gravemente danneggiate, mentre i centri legati alle funzioni motorie primarie e sensoriali sono relativamente risparmiati. Uno studio ha trovato che le regioni del cervello che si attivano quando la nostra mente vaga, il cosiddetto stato predefinito o di riposo, sono le stesse in cui si depositano le prime placche amiloidi. Ma dobbiamo essere cauti nel trarre conclusioni, una mente errante non necessariamente provoca la deposizione di Aβ.
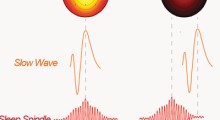
Il sonno è un altro stato elettrico del cervello che è sempre più riconosciuto come un fattore di sviluppo del MA. I livelli sia di Aβ che di tau fluttuano durante il normale ciclo sonno-veglia, e la privazione del sonno aumenta acutamente la produzione di Aβ e diminuisce la sua eliminazione. Il sonno profondo evoca onde ritmiche di liquido cerebrospinale che possono servire a espellere le tossine, come l'Aβ, dal cervello. Purtroppo, questo tipo di sonno diminuisce con l'invecchiamento. Questa osservazione potrebbe stimolare il lavoro sugli approcci farmacologici volti a ristabilire specificamente il sonno profondo.
Idee condivise
Queste aree di ricerca non sono l'alfa e l'omega di un ringiovanito programma di scienza di MA. Ce ne sono certamente di più. Ma questi cinque percorsi si intrecciano e, come la biologia stessa, possono essere indagati in molti modi che si alimentano tra loro. Una speranza che ho è che, come la scienza di base riempie le informazioni mancanti, in particolare le informazioni quantitative, i modellatori computazionali e i teorici entreranno in gioco per aiutare a prevedere l'impatto della patologia del MA sui circuiti cerebrali e sui percorsi cellulari.
Mi piacerebbe anche vedere queste direzioni di ricerca suggerire agli investigatori di pensare collettivamente e in modo sistematico e di condividere le loro idee in modo costruttivo. Questo è il modo con cui possiamo arrivare insieme a spingere indietro la nostra ignoranza su questa terribile malattia.
Fonte: Kenneth S. Kosik, professore di neurologia e ricercatore dell'Università della California di Santa Barbara
Pubblicato in Scientific American (> English text) - Traduzione di Franco Pellizzari.
Copyright: Tutti i diritti di eventuali testi o marchi citati nell'articolo sono riservati ai rispettivi proprietari.
Liberatoria: Questo articolo non propone terapie o diete; per qualsiasi modifica della propria cura o regime alimentare si consiglia di rivolgersi a un medico o dietologo. Il contenuto non rappresenta necessariamente l'opinione dell'Associazione Alzheimer onlus di Riese Pio X ma solo quella dell'autore citato come "Fonte". I siti terzi raggiungibili da eventuali collegamenti contenuti nell'articolo e/o dagli annunci pubblicitari sono completamente estranei all'Associazione, il loro accesso e uso è a discrezione dell'utente. Liberatoria completa qui.
Nota: L'articolo potrebbe riferire risultati di ricerche mediche, psicologiche, scientifiche o sportive che riflettono lo stato delle conoscenze raggiunte fino alla data della loro pubblicazione.