La conformazione dei mitocondri, le centrali energetiche delle cellule, coinvolti nella ipotesi 'neurodegenerazione Alu' derivante dalla precedente ipotesi 'cascata mitocondriale'..L'ultima tornata di test falliti dei farmaci per l'Alzheimer sta inducendo i ricercatori a mettere in discussione l'approccio dominante per combattere la malattia, che vuole impedire l'accumulo nel cervello della proteina appiccicosa chiamata amiloide.
La conformazione dei mitocondri, le centrali energetiche delle cellule, coinvolti nella ipotesi 'neurodegenerazione Alu' derivante dalla precedente ipotesi 'cascata mitocondriale'..L'ultima tornata di test falliti dei farmaci per l'Alzheimer sta inducendo i ricercatori a mettere in discussione l'approccio dominante per combattere la malattia, che vuole impedire l'accumulo nel cervello della proteina appiccicosa chiamata amiloide.
Degli scienziati della Duke University hanno identificato un meccanismo nel macchinario molecolare della cellula che potrebbe aiutare a spiegare come i neuroni cominciano a vacillare nelle fasi iniziali dell'Alzheimer, anche prima della comparsa dei grumi amiloidi.
Questo ripensamento del processo dell'Alzheimer è centrato su geni umani cruciali per il corretto funzionamento dei mitocondri (le fabbriche di energia della cellula), che sono pieno di pezzi mobili di DNA chiamati «elementi Alu». Se questi «jumping genes» [geni saltellanti] perdono i loro normali controlli quando la persona invecchia, potrebbero iniziare a devastare il macchinario che fornisce energia alle cellule cerebrali, portando ad una perdita di neuroni e, infine, alla demenza, dicono i ricercatori.
E se questa «ipotesi neurodegenerazione Alu» si rivelerà valida, potrebbe aiutare a identificare più presto le persone a rischio, prima che sviluppino i sintomi, o puntare a nuovi modi per ritardare l'insorgenza o rallentare la progressione della malattia, ha detto il co-autore Peter Larsen, ricercatore senior del laboratorio del professore di biologia Anne Yoder alla Duke.
L'idea dominante che guida la ricerca di Alzheimer da 25 anni è che la malattia risulta dall'accumulo anomalo di placche amiloidi dure e cerose nelle aree del cervello che controllano la memoria. Ma tutte le sperimentazioni di farmaci anti-amiloide sono fallite, portando alcuni ricercatori a teorizzare che l'accumulo di amiloide è una conseguenza della malattia, non una causa.
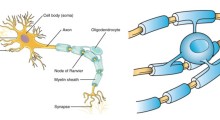
Lo studio della Duke si basa su un'ipotesi alternativa. Proposta la prima volta nel 2004, l'ipotesi «cascata mitocondriale» postula che sono i cambiamenti nelle centrali energetiche cellulari la causa della morte dei neuroni, non l'accumulo di amiloide. Come la maggior parte delle cellule umane, i neuroni si basano su mitocondri per rimanere in buona salute. Ma a differenza delle altre cellule, la maggior parte dei neuroni smette di dividersi dopo la nascita, per cui non possono essere sostituiti se si danneggiano.
Nei pazienti di Alzheimer, dice l'ipotesi, sono i mitocondri dei neuroni a smettere di funzionare correttamente. Di conseguenza non sono in grado di generare energia sufficiente per i neuroni, che patiscono la fame e muoiono, e non c'è modo di sostituirli. Ma purtroppo non si sa come e perché i mitocondri nei neuroni declinano con l'età.
La maggior parte delle proteine mitocondriali sono codificate da geni nel nucleo delle cellule, prima di raggiungere la loro destinazione finale nei mitocondri. Nel 2009, Allen Roses, neurologo della Duke e coautore dello studio (ora deceduto), ha identificato un'area non codificante di lunghezza variabile in un gene chiamato TOMM40. Rose e il suo team hanno scoperto che la lunghezza di questa regione può aiutare a prevedere il rischio di Alzheimer di una persona e l'età di insorgenza.
Larsen si è chiesto se la variazione di lunghezza nel TOMM40 era solo una parte dell'equazione. Ha analizzato la regione del gene corrispondente nei topi lemuri grigi, primati grandi come una tazza da tè, che con l'età sviluppano placche cerebrali amiloidi e altri sintomi di tipo Alzheimer. Ha scoperto che solo nei topi lemuri, ma non in altre specie di lemuri, la regione è piena di tratti brevi di DNA chiamati Alu.
Gli Alu, presenti solo nei primati, appartengono ad una famiglia di retrotrasposoni o «geni che saltano», che si copiano e si incollano in nuovi punti del genoma. Se le copie di Alu presenti all'interno del gene TOMM40 interferiscono in qualche modo con il percorso dal gene alla proteina, ha ragionato Larsen, questo potrebbe contribuire a spiegare perché i mitocondri nelle cellule nervose smettono di funzionare.
"Gli elementi Alu sono una spada a doppio taglio", ha detto Larsen. Prima respinti come spazzatura inutile del DNA, ora ne è riconosciuto il contributo alla diversità e alla complessità del cervello umano. "Essi possono dare funzioni nuove e benefiche dei geni", ha detto Larsen. "Hanno aiutato l'evoluzione delle funzioni cognitive superiori degli esseri umani, ma forse a costo della vulnerabilità del neurone, che aumenta con l'età".
Quando i ricercatori hanno cercato in tutto il genoma umano, hanno scoperto che gli Alu avevano più probabilità di essere in agguato dentro e intorno ai geni essenziali per i mitocondri, rispetto ad altri geni codificanti le proteine. Gli Alu di solito sono tenuti sotto controllo da gruppi di atomi chiamati «gruppi metilici» che si attaccano alla parte esterna del DNA e ne spengono la capacità di saltare o di spegnere/accendere i geni. Ma nel cervello che invecchia, cambiano i modelli di metilazione del DNA, risvegliando alcune copie di Alu, secondo Larsen.
Il gene TOMM40 codifica una proteina di forma cilindrica nella membrana esterna dei mitocondri che forma un canale per far entrare le molecole, compreso il precursore dell'amiloide. Larsen ha usato la modellazione 3D per mostrare che inserti di Alu nel gene TOMM40 potrebbero indurre la proteina canale che esso codifica a ripiegarsi nella forma sbagliata, con l'effetto di intasare i macchinari di importazione dei mitocondri, che smettono di funzionare.
E' probabile che tali processi siano attivati prima dell'accumulo di amiloide, e quindi possono farci puntare a farmaci nuovi o riproposti per un intervento più precoce, ha detto il co-autore Michael Lutz, professore assistente di neurologia alla Duke.
Il gene TOMM40 è un esempio, dicono i ricercatori, ma se gli Alu disturbano altri geni mitocondriali, lo stesso meccanismo di base potrebbe contribuire a spiegare le fasi iniziali anche di altre malattie neurodegenerative, tra cui il Parkinson, il morbo di Huntington e la sclerosi laterale amiotrofica (SLA).
I ricercatori descrivono l'ipotesi «neurodegenerazione Alu» in un documento pubblicato online su Alzheimer's & Dementia: The Journal of the Alzheimer's Association.
"Dobbiamo cominciare a pensare in modo nuovo se vogliamo curare le malattie neurologiche come l'Alzheimer", ha detto Larsen, che ha depositato un brevetto provvisorio per conservare le funzioni mitocondriali tenendo sotto controllo gli Alu.
Fonte: Duke University via EurekAlert! (> English text) - Traduzione di Franco Pellizzari.
Riferimenti: Peter A. Larsen, Michael W. Lutz, Kelsie E. Hunnicutt, Mirta Mihovilovic, Ann M. Saunders, Anne D. Yoder, Allen D. Roses. The Alu neurodegeneration hypothesis: A primate-specific mechanism for neuronal transcription noise, mitochondrial dysfunction, and manifestation of neurodegenerative disease. Alzheimer's & Dementia: The Journal of the Alzheimer's Association, Published online: February 24, 2017. DOI: http://dx.doi.org/10.1016/j.jalz.2017.01.017
Copyright: Tutti i diritti di eventuali testi o marchi citati nell'articolo sono riservati ai rispettivi proprietari.
Liberatoria: Questo articolo non propone terapie o diete; per qualsiasi modifica della propria cura o regime alimentare si consiglia di rivolgersi a un medico o dietologo. Il contenuto non dipende da, nè impegna l'Associazione Alzheimer onlus di Riese Pio X. I siti terzi raggiungibili da eventuali links contenuti nell'articolo e/o dagli annunci pubblicitari sono completamente estranei all'Associazione, il loro accesso e uso è a discrezione dell'utente. Liberatoria completa qui.
Nota: L'articolo potrebbe riferire risultati di ricerche mediche, psicologiche, scientifiche o sportive che riflettono lo stato delle conoscenze raggiunte fino alla data della loro pubblicazione.